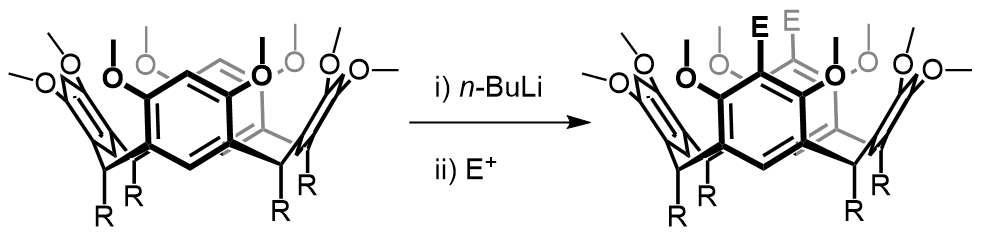RESEARCH
Organic Synthesis with a focus on calixarenes
OVERVIEW:
 Although the Arnott group has a general interest in synthetic organic chemistry, the research is currently directed towards the areas of calixarene and resorcinarene chemistry. Both these structures provide a platform for our strategies to elaborate and expand on the chemistry of these molecules who have found applications in
Although the Arnott group has a general interest in synthetic organic chemistry, the research is currently directed towards the areas of calixarene and resorcinarene chemistry. Both these structures provide a platform for our strategies to elaborate and expand on the chemistry of these molecules who have found applications in  many different areas such as host-guest chemistry, molecular self-assembly, nanotechnology and many other areas. If one has never been exposed to these molecules, it can be surprising at first, since they have a definite 3D shape and cavity, but this is ultimately where the interest in these molecules come from. One advantage is that they are easy to synthesise and can be obtained in multi-gram quantities by even the most inexperienced lab student!
many different areas such as host-guest chemistry, molecular self-assembly, nanotechnology and many other areas. If one has never been exposed to these molecules, it can be surprising at first, since they have a definite 3D shape and cavity, but this is ultimately where the interest in these molecules come from. One advantage is that they are easy to synthesise and can be obtained in multi-gram quantities by even the most inexperienced lab student!
CALIXARENES:
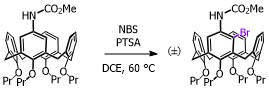
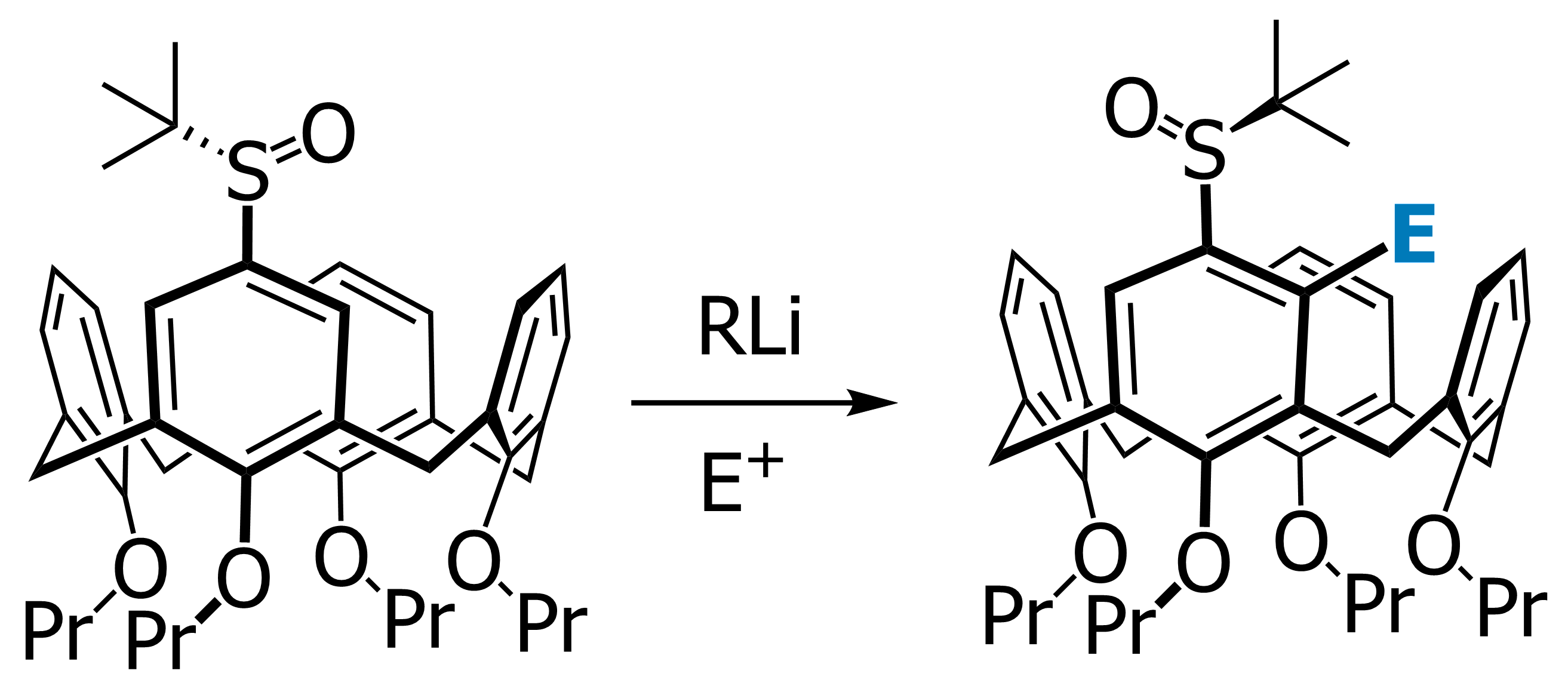
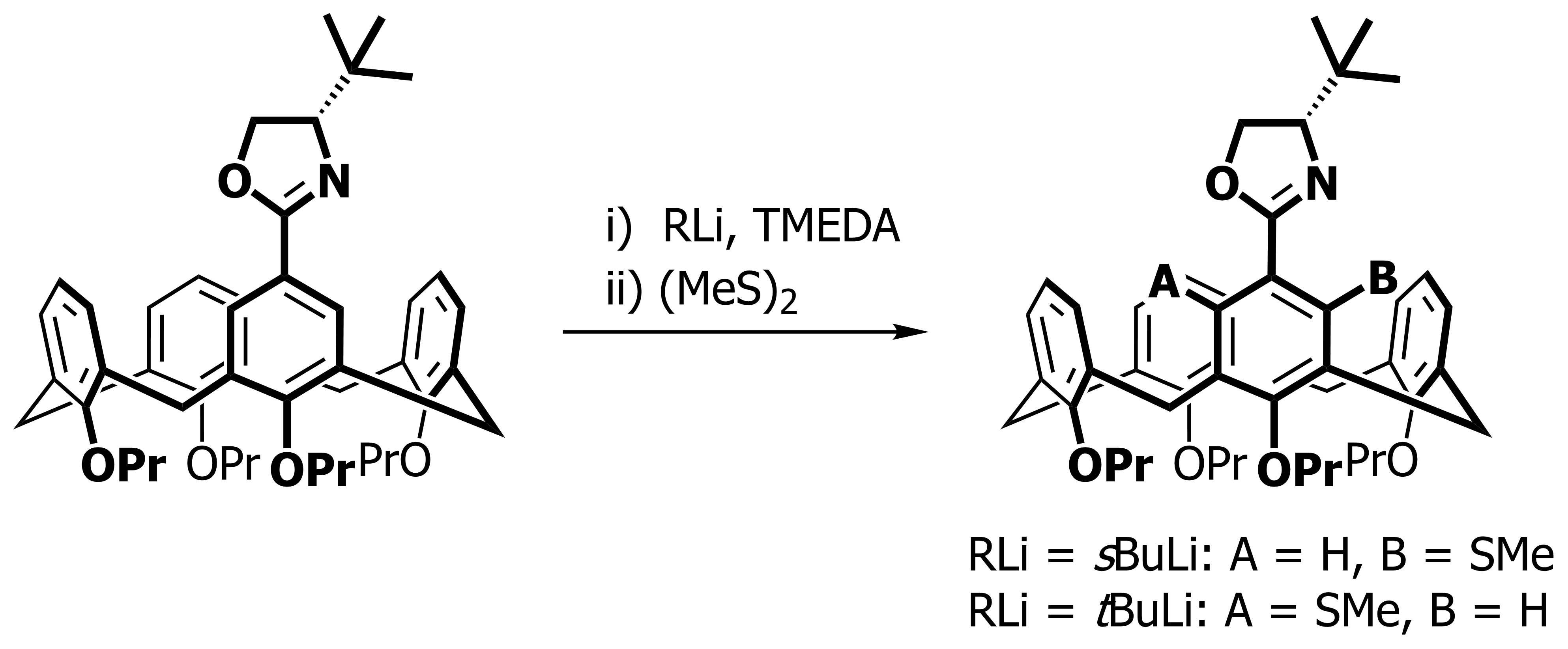
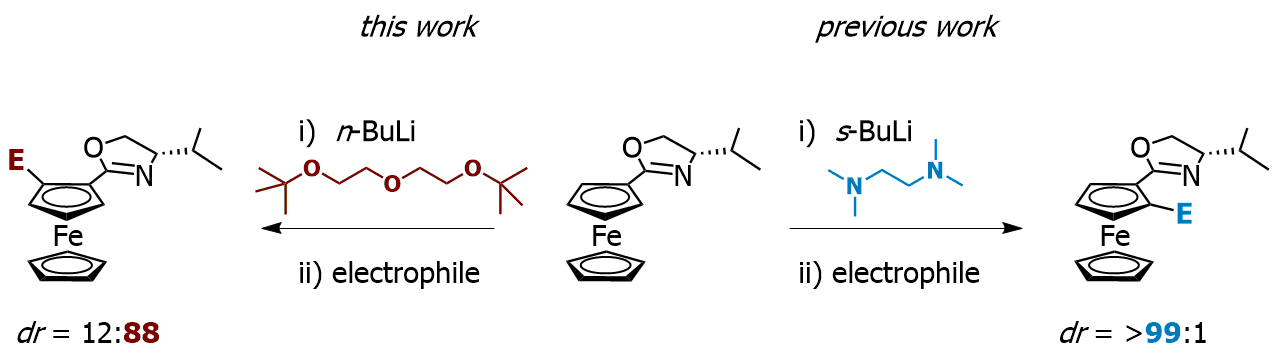
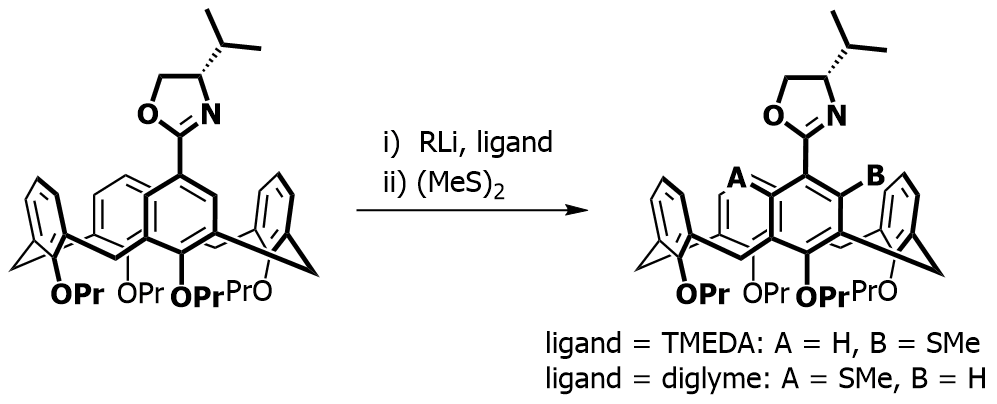
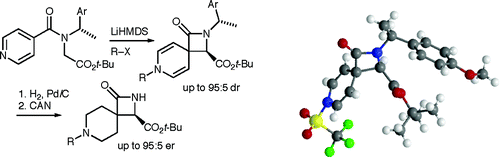
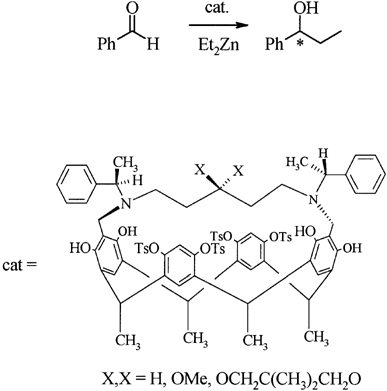
 Our focus with these molecules is to develop the chemistry around inherent chirality. This can be a difficult concept ‘see’, but if you consider the picture on the left, you should realise that the two calixarenes are mirror images of each other and that they are non-superposable. This meets all the criteria for chirality, even though there are no typical sp3 chiral carbon atoms. This feature has been known in calixarenes for many years, but precious few methods for their synthesis exist. Indeed we were the first group to come up with a method that enables the synthesis of meta-substituted inherently chiral calixarenes. The method (see scheme below) relies on an ortholithiation strategy using a chiral oxazoline. One of the unusual results was that the diastereoselectivity of the reaction could be switched by simply changing an achiral additive in the reaction.
Our focus with these molecules is to develop the chemistry around inherent chirality. This can be a difficult concept ‘see’, but if you consider the picture on the left, you should realise that the two calixarenes are mirror images of each other and that they are non-superposable. This meets all the criteria for chirality, even though there are no typical sp3 chiral carbon atoms. This feature has been known in calixarenes for many years, but precious few methods for their synthesis exist. Indeed we were the first group to come up with a method that enables the synthesis of meta-substituted inherently chiral calixarenes. The method (see scheme below) relies on an ortholithiation strategy using a chiral oxazoline. One of the unusual results was that the diastereoselectivity of the reaction could be switched by simply changing an achiral additive in the reaction.

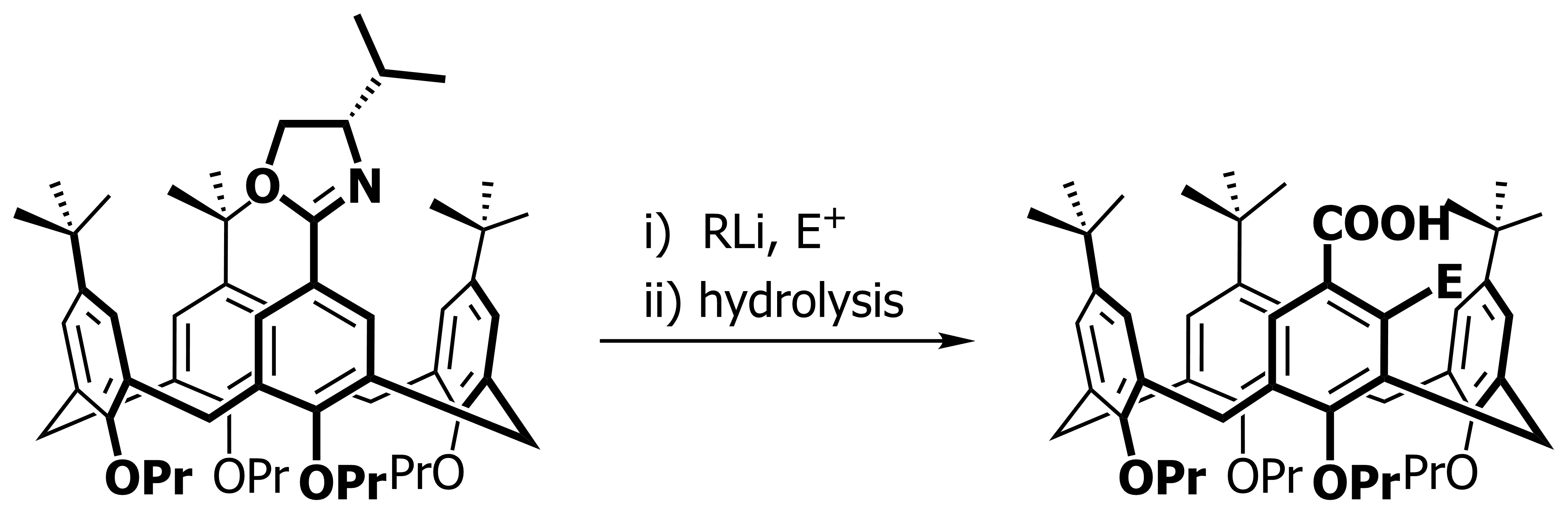
Our current work in this area seeks to expand the method in a number of directions. The first is to look at other methods that may also be used to synthesise inherently chiral calixarenes, such as ortholithiation directed by chiral sulfoxides. Secondly we are interested in extending the method to the other rings of the calixarene, and thirdly we are looking at applying the method to synthesise specific targets that could act as ligands in asymmetric catalytic processes.
RESORCINARENES:
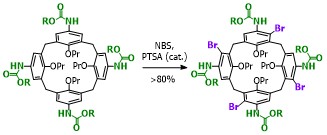
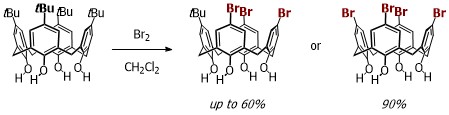
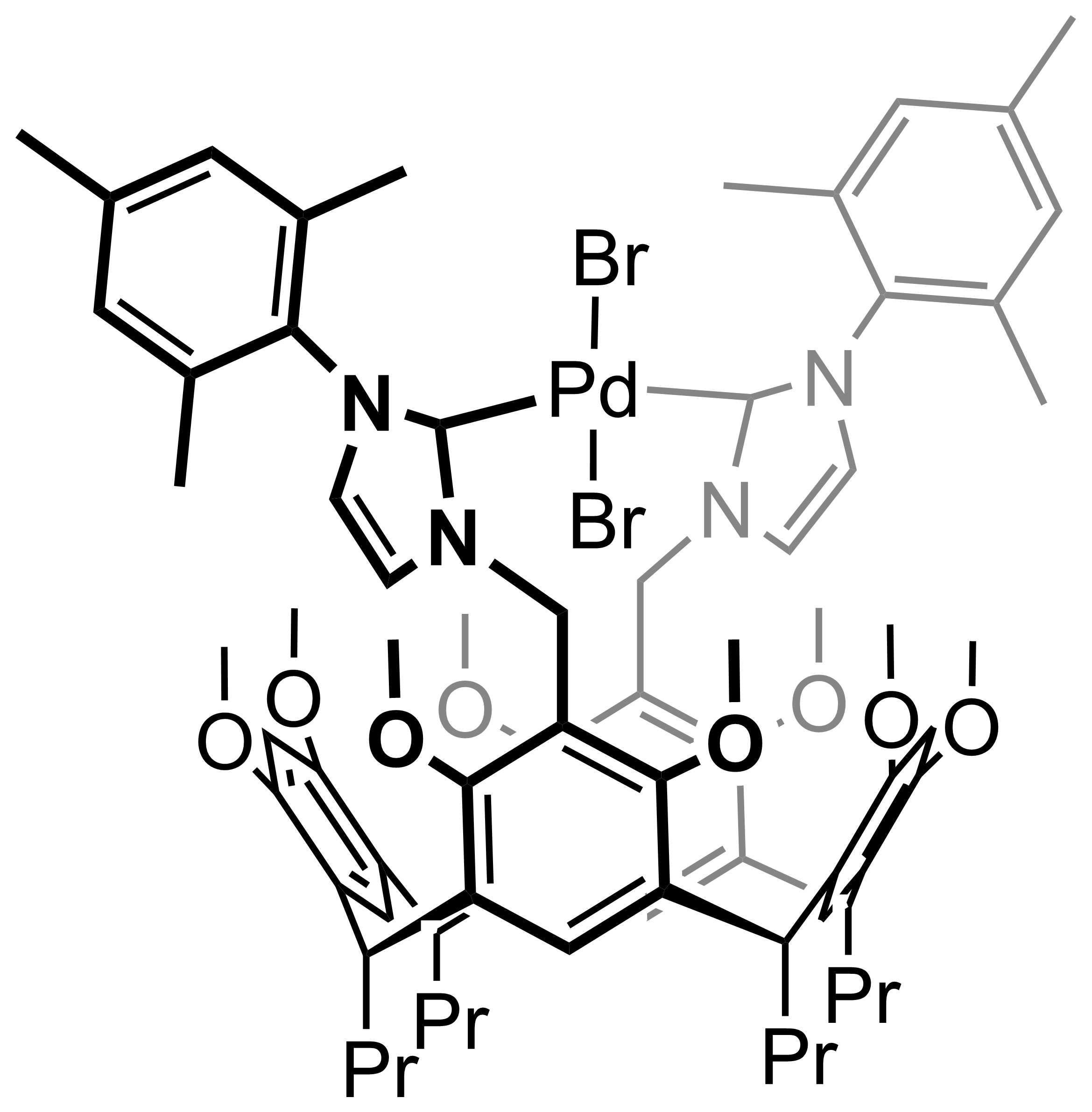
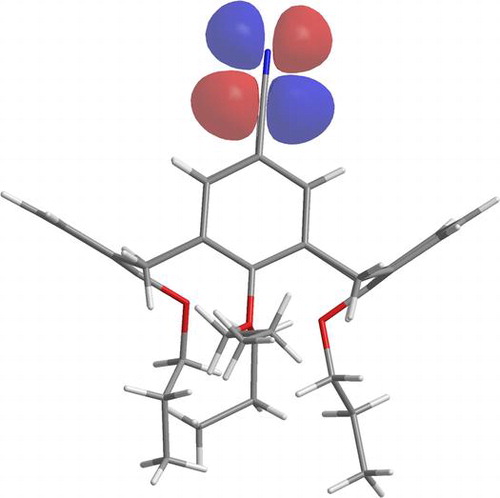
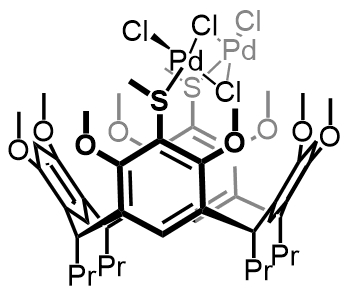
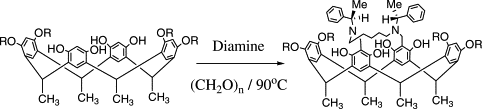
 Our work in resorcinarenes has its roots in the PhD work of Gareth Arnott. Like calixarenes, these too can have inherently chiral isomers, but our work for the moment has been on methods for the selective distal functionalization of these molecules. By distal, we mean that some functionality has been introduced on the aromatic rings that are opposite each other in the resorcinarene. We have been interested in whether suitably functionalised versions of these molecules may act as bidentate ligands in transition-metal catalysed processes. So far we have been successful in developing a method for the synthesis of distal functionalised resorcinarenes and we have recently been exploring N-heterocyclic carbene analogues.
Our work in resorcinarenes has its roots in the PhD work of Gareth Arnott. Like calixarenes, these too can have inherently chiral isomers, but our work for the moment has been on methods for the selective distal functionalization of these molecules. By distal, we mean that some functionality has been introduced on the aromatic rings that are opposite each other in the resorcinarene. We have been interested in whether suitably functionalised versions of these molecules may act as bidentate ligands in transition-metal catalysed processes. So far we have been successful in developing a method for the synthesis of distal functionalised resorcinarenes and we have recently been exploring N-heterocyclic carbene analogues.
OTHER PROJECTS / COLLABORATIONS:
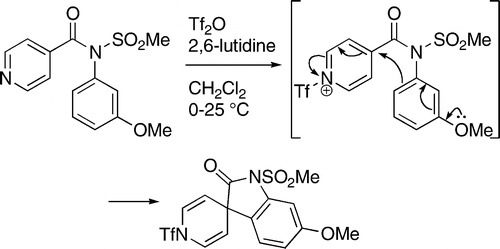
Within the department of chemistry and polymer science, we are involved, or have been involved, in a number of collaborative projects, such as dithiadiazolyl chemistry with Delia Haynes, polymer functionalization with Bert Klumperman and Peter Mallon, and organic synthesis with Willem van Otterlo.
GROUP MEMBERS
Current members of the Arnott group

Trégen Snayer
M.Sc. (2018—2019)
Ph.D. (2020—)
ALUMNI
Past members of the Arnott group

Asslly Mafaune
M.Sc. (2015—2017)
Ph.D. (2018—2022)

Chris Jurisch
M.Sc. (2016—2017)
Ph.D. (2018—2021)

Dr Luke Hodson
Postdoc (2019)

Babak Eghbali
Ph.D. (2013—2019 )

Kevin Visagie
M.Sc. (2017—2019)

Dr Sritama Bose
Postdoc (2016—2017)

Ashlyn Bhana
M.Sc. (2015—2017)

Dr Dominic Castell
Ph.D. (2013—2016)

Dr Lonwabo Ngodwana
M.Sc. (2010—2012)
Ph.D. (2013—2016)

Dr Irene Tosi
Postdoc (2015)

Dr Dewald Kleinhans
M.Sc. (2008—2009)
Ph.D. (2010—2015)

Lutho Siko
NRF Intern (2014—2015)

Nwabisa Wali
NRF Intern (2014)

Laura van Laeren
M.Sc. (2011—2013)

Malcolm Applewhite
M.Sc. (2010—2011)

Ntlama Lesotho
Ph.D. (2008— )

Dr Simon Herbert
Ph.D. (2008—2011)